医疗器械单一审核计划 (MDSAP) 涵盖了 ISO 13485:2016 标准以及医疗器械出口至五个国家(澳大利亚、巴西、加拿大、日本和美国)的法规要求。请参见 MDSAP 服务页面,详细了解拥有 MDSAP 证书带来的诸多优势。在本文中,我们将探讨获得 MDSAP 证书所需采取的关键步骤。
为了获得 MDSAP 证书,制造商的质量管理体系 (QMS) 必须符合 ISO 13485:2016 标准的适用要求。此外,根据制造商销售或拟销售医疗器械的国家,其 QMS 必须包含/整合各个 MDSAP 国家的医疗器械法规和要求。尽管 MDSAP 覆盖了五个不同的成员国,但在申请 MDSAP 认证时,QMS 只需满足制造商适用的成员国的要求。因此,如果制造商最初仅计划在美国和澳大利亚销售其设备,那么在首次注册审核期间,其 QMS 只需符合美国食品药品监督管理局 (FDA) 和澳大利亚治疗用品管理局 (TGA) 的适用要求。如果制造商计划未来在其他 MDSAP 国家进行市场推广,则需要升级 QMS 以符合其他国家的要求,并相应地扩大证书范围。
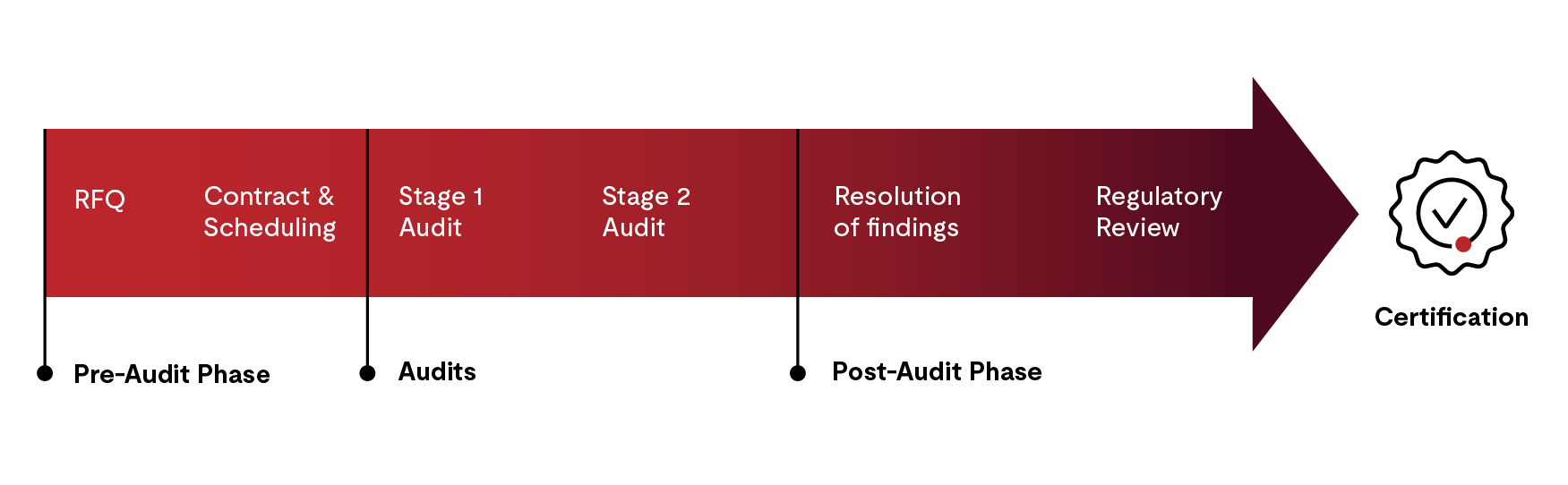
认证过程分为以下四个步骤:
- 预审阶段,以出具报价、签订合同并安排审核
- 第一阶段和第二阶段审核
- 审核后活动
- 证书发放
为了顺利完成每一个步骤,制造商需要进行充分准备,并获得管理层坚定的支持。
预审阶段
在签署合同之前,客户需要提供有关其组织和设施、计划销售的医疗器械产品、员工人数以及其他相关信息。UL Solutions 将根据初步信息提供报价,并在签署合同之前回答与报价相关的任何问题,包括首次注册审核的预计时间表。如果客户接受报价,则客户将与审核机构 (AO) 签署合同,并安排注册审核事宜。
第一阶段审核
第一阶段审核 (S1) 的主要目标是评估客户的 QMS 是否已准备就绪,可以满足 MDSAP 要求。审核重点关注定义 QMS 的书面程序(标准操作程序 (SOP) 和相关文件)。SOP 应涵盖 ISO 13485:2016 和《MDSAP 审核方法》文件 (MDSAP AU P0002) 中规定的适用流程。《MDSAP 审核方法》文件已在 MDSAP 文件网站上公开发布。客户应在 QMS 中体现对 ISO 13485:2016 标准和 《MDSAP 审核方法》指导方针的充分理解。第一阶段审核可以采取现场或远程方式进行。建议有关 SOP 都准备电子版,以便远程查看。在审核过程中,审核机构将重点审核质量手册,因为这通常是 QMS 的核心文件。
值得注意的是,在某些 MDSAP 成员国,外国制造商在首次市场准入流程中,必须寻求当地实体的协助,而在其他国家则不做强制要求。
- 在澳大利亚,外国制造商必须委托当地机构作为其代表,并向 TGA 注册其医疗器械。该代表称为澳大利亚担保人。
- 巴西境外的外国制造商必须设立一个当地实体作为巴西注册持有人 (BRH),才能获得巴西的市场准入许可。
- 在日本销售医疗器械时,制造商必须委任当地代表(称为销售授权持有人 (MAH)),代表制造商与卫生部沟通。
- 在加拿大销售医疗器械设备(2 类及以上)的制造商必须持有 MDSAP 证书。在 MDSAP 的五个成员国中,目前只有加拿大强制执行 MDSAP。对于加拿大卫生部认定的 1 类器械,不需要获得 MDSAP 证书。在加拿大,外国制造商可以选择与加拿大的进口商合作,但这并非强制性要求。制造商可以直接向加拿大卫生部申请医疗器械许可证。制造商可以聘请第三方监管联络人,由其协助与加拿大卫生部进行沟通,但这并非强制性要求。
- 在美国,医疗器械设备制造商可以直接与 FDA 沟通。
在 S1 审核阶段,制造商必须证明对每个适用监管机构的基本要求具备相应了解。如果在 S1 审核期间发现任何缺漏,审核机构将告知客户,并要求其在第二阶段审核 (S2) 之前解决。发现缺漏可能会导致两种结果:
- 认定客户的 QMS 尚未做好满足 MDSAP 要求的准备,建议重新进行 S1 审核;
- 认定所发现的缺漏相对较小,建议进行下一步的 S2 审核。根据缺漏程度的不同,S2 审核可能安排在 S1 审核后的一个至六个月内进行。如果间隔时间超过 6 个月,则需要再次进行 S1 审核。
第二阶段审核
第二阶段 MDSAP 审核(S2 审核)在现场进行,通常持续数天。在签署合同之前,审核机构会事先通知制造商所需的审核天数。该审核采用 MDSAP 审核方法模型,要求按照以下结构对 QMS 的不同方面进行评估:
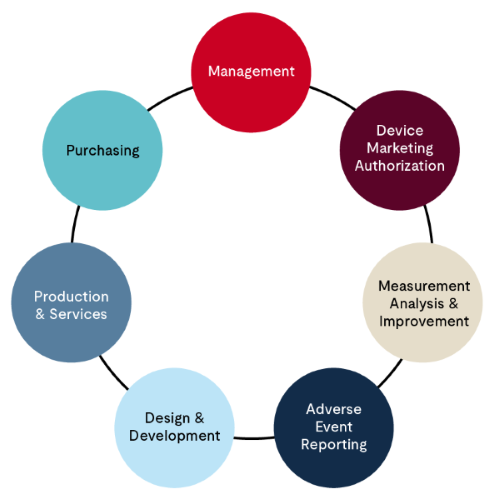
- 管理流程——共分为 11 项任务,主要关注 QMS 的整体情况,包括但不限于组织结构、管理层承诺、管理层评审、文件控制、人力资源和能力等。质量手册是评估的重点。
- 器械市场准入流程——主要涉及向相关监管机构申请设施注册和器械许可的流程。该流程分为三项任务,通常由申请 MDSAP 认证的组织内的监管专家负责管理。
- 测量、分析和改进——顾名思义,主要涉及监控 QMS 的运作情况,并采取相应的改进措施。投诉处理、CAPA(纠正和预防措施)、内部审核流程、不合格材料和流程以及上市后监督构成了这一流程的核心要素。该流程通常由申请 MDSAP 认证的组织内的质量保证经理负责管理。
- 不良事件报告、召回和警示通知——根据 MDSAP 审核方法,该流程中有三个任务需要评估。该流程通常由申请 MDSAP 认证的组织内的法规事务团队负责管理。
- 设计和开发流程——根据 MDSAP 审核方法,该流程包含 16 项评估任务,涵盖了新器械设计和开发过程中从规划、设计转移到制造的有关环节。还包括设计变更流程。
- 生产与服务控制流程——MDSAP 审核方法建议执行 29 项评估任务,以涵盖生产和服务控制过程。该部分涵盖了器械生产的所有环节,包括但不限于基础设施、工作环境、生产流程验证、生产质量控制测试、存储、识别和可追溯性、不合格材料处理以及销售等。
审核后活动
审核后活动主要包括以下两项活动:
- 制造商对审核过程中发现的不符合项进行整改,以及
- 审核机构对审核结果进行内部监管审查。
在 S1 审核中发现的不符合项必须在 S2 审核中加以解决、消除并得到验证。在 S2 审核过程中,如果发现不符合项,则需根据 MDSAP 的要求将其划分为 1 到 5 级。MDSAP 采用了 GHTF/SG3/N19:2012 分级方案的基本概念。本 GHTF 源文件的发布日期早于 ISO 13485:2016,因此,文件中随处可见对 ISO 13485:2003 的引用。不过,其中提到的分级方案的原则和概念仍然适用。在发布 MDSAP 证书之前,必须解决 4 级和 5 级的不符合项,或被归类为 ISO 13485 主要问题的不符合项。
审核小组将审核结果提交给内部监管审查小组。根据本次审查的结果,证书决策者将对审核进行评估,以确定是否达到成功标准,并决定是否颁发 MDSAP 证书,然后将此决定通知制造商。根据 MDSAP 政策的要求,最终审核报告包也将提交至 MDSAP REPS 数据库。
MDSAP 证书必须符合 MDSAP AU P0026 的要求,至少应包含以下内容:合法制造商的名称和地址、认证范围、适用的司法管辖区、认证周期的起始日期、有效期以及证书到期日期。通常情况下,证书的有效期为三年。
为什么选择 UL Solutions 来进行 MDSAP 审核?
UL Solutions 是全球应用安全科学专家。作为 UKAS 认可的 ISO 17021 注册机构,UL Solutions 可为多个项目提供 QMS 注册服务。经 UKAS 认可,我们可提供 ISO 13485 审核以及 ISO 13485 和 ISO 9001 的联合审核服务。
我们的审核人员是 UL Solutions 的全职员工,可在 QMS 的生命周期内提供一致的审核体验和支持。
我们拥有经验丰富的监管审核员和评估员,他们来自于超过 1.5 万名富有使命感的员工群体,分布于 40 多个国家,并为来自 100 多个国家的客户提供服务。
UL 认证标志在全球范围内传递信任,也反映了我们对推进安全使命的坚定承诺。
作者简介
Chira Deka 是 UL Solutions MDSAP 项目的项目经理。与此同时,他还担任 ISO 13485、ISO 9001 和 MDSAP 认证计划的区域首席审核员、证书决策者和首席审核员,MDD 和 IVDD 计划的首席审核员,以及 UKCA 认证计划的审核员。他是 IVD 技术方面的技术专家,在该领域拥有多项专利。此外,他曾是美国国立卫生研究院 (NIH) SBIR 技术评审小组(微生物学)的成员,并且曾获得 NIH 和美国商务部 ATP 研究 IVD 和生物技术方面的资助。



X
与我们的销售团队取得联系
感谢您关注我们的产品和服务。我们将收集一些信息,以便安排合适的人员与您联系。
Please wait…