Medical Device Single Audit Program (MDSAP) encompasses the requirements of ISO 13485:2016 and the medical device regulations of five countries: Australia, Brazil, Canada, Japan, and U.S. combined. Our MDSAP service page covers the benefit of having an MDSAP certificate. In this article, we will discuss key steps needed before an MDSAP certificate can be granted.
In order to obtain an MDSAP certificate, the Quality Management System (QMS) must meet all applicable requirements of the ISO 13485:2016 standard. Additionally, it must incorporate/integrate the medical device regulations and requirements of each MDSAP country where a manufacturer is selling or wishes to sell medical devices. Even though MDSAP covers five different member countries, the QMS is requirement to meet the requirements only of the countries that are applicable to the manufacturer at the time of certifications. Thus, if a manufacturer wishes to initially market its devices only in the U.S. and Australia, the QMS must meet the applicable requirements of only U.S. Food and Drug Administration (FDA) and Australia Therapeutic Goods Administration (TGA) during the initial registration audits. Marketing in additional MDSAP countries in the future will require the QMS to evolve to incorporate the requirements of the additional countries and the certificate scope can be expanded accordingly.
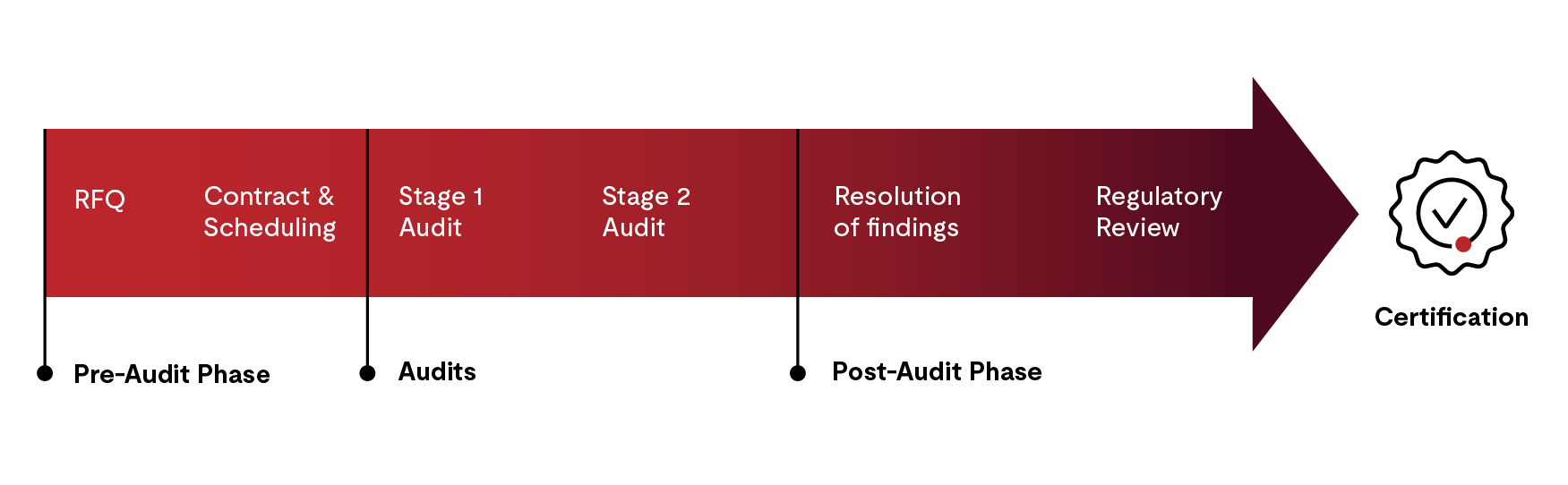
The process of certification comprises four steps:
- Pre-audit phase for quote, contract, and audit scheduling
- Stage 1 and Stage 2 audits
- Post-audit activities
- Certificate issuance
Navigating each step successfully requires careful preparation by the manufacturer and strong management commitment.
Pre-audit phase
Prior to signing a contract, the customer will need to share basic information about the organization and facilities, medical device products it intends to market, headcount, and other details. UL Solutions will provide a quote based on the preliminary information and answer any questions related to the quote, including projected timeframes of the initial registration audits, prior to signing a contract. If the customer accepts the quote, a contract between the customer and the auditing organization (AO) will be signed and the registration audits will be scheduled.
Stage 1 audit
The main purpose of a Stage 1 audit (S1) is to evaluate the customer’s QMS for readiness to meet the MDSAP requirements. Emphasis is given on documented procedures (SOPs and related documents) that define the QMS. SOPs are expected to cover all applicable processes defined in ISO 13485:2016 and the MDSAP Audit Approach document (MDSAP AU P0002). The MDSAP Audit Approach document is publicly available at the MDSAP documents website. A comprehensive understanding of ISO 13485:2016 standard and the guidelines of the MDSAP Audit Approach should be reflected in the QMS. Stage 1 audit can be on-site or remote. It is best to have all SOPs in electronic form so that they can be viewed remotely. Among other things, the Quality Manual of the audited organization will be closely scrutinized during this audit, as it is usually a centerpiece of any QMS.
It should be noted that in certain MDSAP member countries, foreign manufacturers must seek assistance from a local entity in the initial market authorization process, while in some others, it is optional.
- For Australia, a foreign manufacturer must engage an Australia-based party to represent it and register its medical devices with TGA. This representative is called an Australian sponsor.
- A foreign manufacturer outside Brazil must have a local entity to serve as the Brazilian registration holder (BRH) to obtain marketing authorization in Brazil.
- For marketing medical devices in Japan, a manufacturer must have a local representative, called a marketing authorization holder (MAH) to communicate with the Ministry of Health on the manufacturer’s behalf.
- To market medical devices (Class 2 and above) in Canada, a manufacturer must have an MDSAP certificate. Of the five member countries of MDSAP, currently MDSAP is mandatory only in Canada. For a Health Canada Class 1 device, an MDSAP Certificate is not required. In Canada, a foreign manufacturer may engage an importer based in Canada, but it is not mandatory. A manufacturer can apply for a medical device license in Canada directly with Health Canada. A manufacturer can engage a third-party regulatory correspondent to assist in interactions with Health Canada, but it is not mandatory.
- In the U.S., a medical device manufacturer can communicate directly with the FDA.
Awareness of the basic requirements from each of the applicable regulatory agencies must be demonstrated during an S1 audit. Gaps identified during the S1 audit will be communicated to the customer and must be addressed before the Stage 2 audit (S2). Gaps may lead to two possible results:
- Determination that customer QMS is not ready for MDSAP and a new S1 audit may be recommended, or,
- Determination that the gaps identified are relatively minor in nature and therefore, an S2 audit is recommended as the next step. Depending on the degree of the gap, the S2 audit may be scheduled one to six months from the S1 audit. If the gap is greater than six months, a new S1 audit will be required again.
Stage 2 audit
Stage 2 MDSAP audit (S2 audit) is a conducted on-site and typically spans several days. The number of audit days would be already provided to the manufacturer prior to the signing of the contract. The audit is conducted using the MDSAP audit approach model, which requires the following structure to evaluate different aspects of the QMS, as follows:
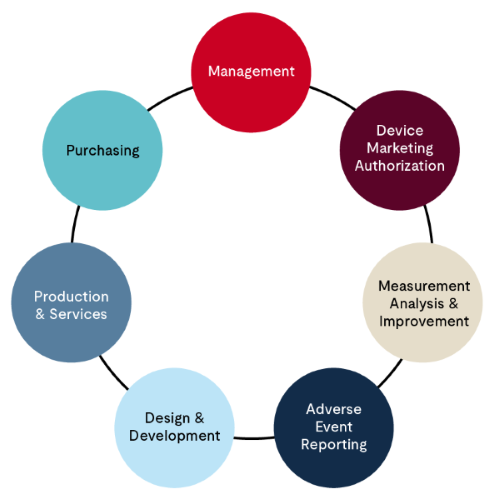
- Management process – Divided into 11 tasks, this focuses on the overview of the QMS, including but not limited to, organizational structure, management commitment, management review, document control, human resources and competency. The Quality Manual is key to this evaluation.
- Device marketing authorization process – This is focused on facility registrations with applicable regulatory agencies and device licensing processes. This is divided into three tasks. These processes are often managed by a regulatory expert within the organization seeking MDSAP certification.
- Measurement, analysis, and improvement – As the name implies, it is all about monitoring of the functioning of the QMS and initiatives needed for improvement. Complaint handling , CAPA, Internal audits process, nonconforming materials and processes, and post-market surveillance form the core of this process. These processes are often managed by a QA manager within the organization seeking MDSAP certification.
- Adverse event reporting, recalls, and advisory notices – There are three tasks within this process that are evaluated under MDSAP audit approach. These processes are often managed by a regulatory affairs team within the organization seeking MDSAP certification.
- Design and development process – There are 16 tasks for this process in the MDSAP audit approach that cover everything from planning to design transfer to manufacturing when a new device is designed and developed. It also includes design change processes.
- Production and services control process – In the MDSAP Audit Approach, there are 29 Tasks recommended to cover the production and services control processes. This section covers everything that goes into manufacturing devices, including but not limited to, infrastructure, work environment, production process validations, production quality control testing, storage, identification and traceability, handling of nonconforming materials, and sales.
Post-audit activities
Post-audit activities include two main activities:
- Addressing, by the manufacturer, of the nonconformities identified during an audit, and
- Internal regulatory review of the audit results by the auditing organization.
Nonconformities identified during an S1 audit must be addressed, eliminated, and verified at the S2 audit. If nonconformities are identified during an S2 audit, they are graded 1 to 5 according to MDSAP requirements. The basic concept of this grading scheme was adopted by MDSAP from GHTF/SG3/N19:2012. This GHTF source document predates ISO 13485:2016, and as a result, reference to ISO 13485:2003 can be found throughout the document. However, the principle and concept of the grading scheme mentioned there continue to be applicable. Grade 4 and 5 nonconformities or nonconformities classified as ISO 13485 Major findings must be resolved before an MDSAP certificate is released.
The audit team submits the audit results to the internal regulatory review team. Based of the outcome of this review, certificate decision maker will determine if the audit was successful enough to issue the MDSAP certificate and the decision is communicated to the manufacturer. Final audit report package is also submitted to the MDSAP REPS database as required by MDSAP policies.
The MDSAP certificate must follow the requirements of MDSAP AU P0026 and will contain, at minimum, the name and address of the legal manufacturer, scope of the certification, jurisdictions, cycle start date, effective dates and certificate expiry date listed on it. Normally, a certificate is valid for a period of three years.
Why UL Solutions for MDSAP?
UL Solutions is a global leader in applied safety science. As a UKAS-accredited Registrar under ISO 17021, UL Solutions provides QMS registration to multiple programs. Under UKAS, we offer audits to ISO 13485 and combined audits to ISO 13485 and ISO 9001.
All our auditors are full-time UL Solutions employees providing consistent audit experiences and support throughout the life of your QMS.
Our experienced regulatory auditors and assessors are part of our 15,000 mission-driven employees working in more than 40 countries to serve customers in over 100 countries.
The UL Certification Marks serve as a recognized symbol of trust globally and reflect an unwavering commitment to advancing our safety mission.
About the author
Chira Deka works as the program manager for UL Solutions MDSAP Program. He also serves as a Regional Lead Reviewer, Certificate Decision Maker, and Lead Auditor in the ISO 13485, ISO 9001, and MDSAP certification programs, as a lead auditor for the MDD and IVDD programs, and an auditor in the UKCA certification program. He is a technical expert in IVD technologies and holds several patents in this area. He has previously served as a member of the NIH SBIR technology review panel (microbiology) and was an awardee of NIH and U.S. Department of Commerce ATP research IVD and biotechnology grants.




X
Get connected with our sales team
Thanks for your interest in our products and services. Let's collect some information so we can connect you with the right person.
Please wait…
Characters remaining